Nanobiotechnology Project
Nathan Cady, Samuel Chen, Andrea Kao and TJ Zieziulewicz April 9, 2002
Nanobiotechnology ProjectNathan Cady, Samuel Chen, Andrea Kao and TJ Zieziulewicz April 9, 2002 |
I. Introduction
II. Updates and Changes
III. Difficulties
IV. Summary
Team Journal
| I. Introduction: | ||||||||||
| As a group, we have worked
together over the past few months to bring together various portions of
our design into a conceptual biosensor module.
As was outlined in our initial proposal, we intend to create a
miniaturized biosensor that is capable of detecting bacteria and viruses
from a variety of samples, but mainly blood and sputum from human
subjects. As a team, we have
incorporated the strengths and backgrounds of individual team members to
make our design feasible and realistic.
We have worked together through e-mail contact, small group
meetings, large group teleconferences and occasional video conferences.
As is noted in the team journal, we have met frequently and have
been able to work constructively towards the pursuit of our common goal:
a successful project!
|
||||||||||
| II. Updates and Changes: | ||||||||||
| In order to detail the
progress our team has made over the past months, we have decided to report
on individual portions of the biosensor.
Each team member has been responsible for a portion of the sensor
and has subsequently reported on the updates and/or changes that have been
made during our meetings, discussions and individual research.
|
||||||||||
| Pumps: |
Due to the fact that our proposed device requires pumping approximately 10ml of fluid through the device, we determined that we would need some type of pumping system to move this fluid. We considered some of the options presented in the lecture by Scott Stelick, but decided that since the collection of blood and other samples would likely be with needles and syringes, a syringe pump system could be utilized. Alternatively, we could inject the fluid into a reservoir into the device and move this fluid with an on-board pump. After some deliberation about this issue, we decided that the syringe pump would be the easiest means of moving fluids in our device. Syringe pumps are not “miniature” by any stretch of the imagination, so we have been considering the integration of a syringe pump-like system into our device. The most important aspect of a syringe pump is the ability to accurately control the volume of fluid that is being pumped. Therefore, whether the pump is controlled by a worm drive (simple motor and gears) or by a piezoelectric motor, there must be some controller that determines the rate of pumping. Piezoelectric motors might be the easiest to use, especially since they are mechanically simpler than a gear driven syringe pump. In addition, piezoelectric motors often have lower power requirements which would also make them the better choice for our device. The control of the syringe pump is the most complicated design issue and will likely be best integrated with the rest of the control portions of the device. The signal analysis and output must be handled by some kind of computer or microprocessor and therefore we have decided that pump control should also be controlled by the same processor. This would be fairly simple at first, simply integrating all of the control and analysis in a laptop computer, but later designs would likely utilize some kind of integrated microprocessor.
|
|||||||||
| General Nanofabrication: |
We have considered most of the nanofabrication issues involved with our proposed design and have made several changes to the overall design and fabrication. The most significant change to the original design has been the change from a glass to silicon substrate and the integration of the filter into this substrate. Our original proposal had the filter integrated into the PDMS cover of the device. Upon further discussion, however, we decided that the filter needed to be fabricated more accurately than PDMS could offer. The minimal feature size of the filter is approximately 2mm. Although this is possible to construct in silicon and possibly transfer to PDMS, the reliable transfer of the pattern into PDMS is questionable. Therefore, we decided to simply integrate the filter into the solid substrate that will also include the electronics. Since silicon is easier to pattern and etch than glass, we also decided that a silicon base would be used.
The use of a silicon base creates a problem that was initially
avoided by using a glass substrate. Because
we want to evaporate gold electrodes onto the substrate, we have to
somehow insulate the silicon surface to prevent shorting across the wafer.
After consideration of this problem, we decided that it would be
necessary to grow a layer of insulating silicon dioxide on the fabricated
wafer after microfabrication of the filter.
Once this has been done, it would be relatively easy to evaporate
gold electrodes onto the wafer, patterned via lift-off lithography. The following is a stepwise fabrication sequence that we have
come up with: 1. Photolithographic patterning of the filter module (photoresist) 2. Dry etching of the filter module (reactive ion etching) 3. Removal of photoresist. 4. Deposition of silicon dioxide insulating layer (PECVD) 5. Photolithographic patterning of gold electrodes (photoresist). 6. Deposition of gold (evaporation). 7. Lift-off to remove excess gold and photoresist.
8. Possible silicon
nitride deposition step to insulate electrodes This fabrication sequence can be utilized to construct the main base of our device, but the upper capping layer of PDMS must also be fabricated. We have decided to integrate most of the microfluidic portions of the device into the PDMS as it will be easy to make an initial master pattern and simply re-use these molds to make multiple PDMS cap structures. In the consideration of these nanofabrication procedures, we tried to determine what would be the easiest method of constructing such a device, but also what would be the cheapest. Since a device like ours would likely be single-use, it would make sense to make modules that could be mass-produced cheaply. For this reason, PDMS was chosen as a major substrate. PDMS is cheap to use, but also requires much less fabrication, since only a few initial molds are needed to produce multiple PDMS structures. Along these lines, we also considered what portions of the device would be part of the “disposable” structure and what portions would be reused. We felt that the syringe pump, controller and analysis equipment would all be reusable, but that the chip including the microfluidic channels and the active sensor area would be disposable. This creates one difficulty, in that we have to provide a robust connection between the disposable portion of the device and the reusable portion. This will be considered before our final design is presented/submitted.
|
|||||||||
| Sample Characterization: |
In order to better understand the nature of the samples that we have been using, an individual group member has undertaken an initial study to characterize possible samples and some of their inherent properties. Included below is a short summary of some of the information gleaned from this study and with some relevant references: Biological agents have a moderate to high
potential for large-scale dissemination or a heightened general public
awareness that could cause mass public fear and civil disruption. For
example, anthrax has the potential for adverse public health impact with
mass casualties, and requires broad-based public health preparedness
efforts (e.g., improved surveillance and laboratory diagnosis and
stockpiling of specific medications)[1].
A World Health Organization report estimated that 3 days after the
release of 50 kg of anthrax spores along a 2-km line upwind of a city of
500,000 population, 125,000 infections would occur, producing 95,000
deaths[2]. A
brief intervening period of improvement sometimes follows 1 to 3 days of
these prodromal symptoms, but rapid deterioration follows; this second
phase is marked by high fever, dyspnea, stridor, cyanosis, and shock.
Blood smears in the later stages of illness may contain the characteristic
gram-positive spore-forming bacilli. This finding is only likely to occur
late in the course of disease. Confirmation is obtained by culturing B.
anthracis from blood[3].
Blood sample obtain from patients can be directly injected into the
biosensor which has a symmetric array filter design forced out large
particle size over 2 mm.
Other diseases infected by viruses can also be detected by the microchip
with suitable antibody electrodes. Body fluids collection for testing run
includes blood, urine, sputum, nasopharyngeal aspirate and vesicle fluid.
Other samples such as stool, scrapings or biopsy require dissolved and
diluted in medium before injecting to the sensor. [1] Public Health Assessment of Potential Biological Terrorism Agents Lisa
D. Rotz, Ali S. Khan, Scott R. Lillibridge, Stephen M. Ostroff, and
James M. Hughes http://www.cdc.gov/ncidod/EID/vol8no2/01-0164.htm [2] 2. Report of a WHO group of consultants. Health aspects of chemical and biological weapons. Geneva: World Health Organization; 1970. p. 97-9. [3]
Clinical and
Epidemiologic Principles of Anthrax Theodore J. Cieslak and Edward M.
Eitzen, Jr.
|
|||||||||
| Filter Re-design: |
Inspired by several lectures given, we
have envisioned a new filter design, by fabricating layers of filters at a
tilted angle. We believe this
new filter can resolve the clogging problem that many traditional filters
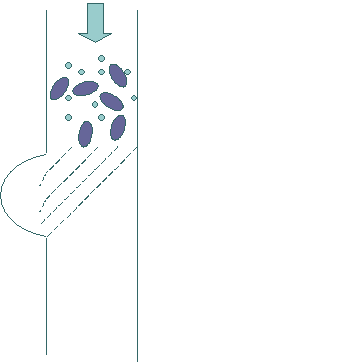
run into. Figure 1 shows the
general the layout of such a filter with pore size 2 mm
wide, 2 mm
high and a 10-20 mm
channel depth. Fig. 1 Fig. 2
As can be seen from Figure 1, this new filter design intends to incorporate both traditional filter design and the novel secondary approach. With the tilted design, the filter prevents the bigger size cells from clogging the entrances to the filtering device. The actual working of this device can be illustrated by the following figures. In Figure 2, we illustrated the intended
design with a sample of blood coming into the filter. The openings of the broken lines are intended to be bigger
than the bacteria/viruses but small than the bigger cells in the blood
sample. In this figure, we
have increased the size of viruses, so that it will be easier to see them. Figure 3 shows the motion of the blood sample through the
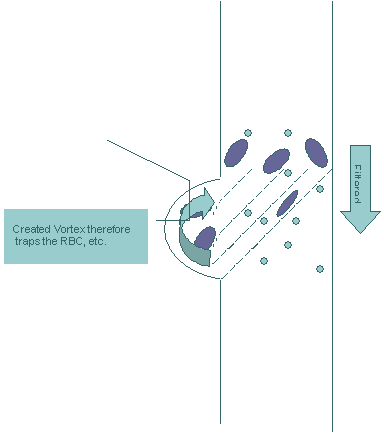
filter. Figure 3
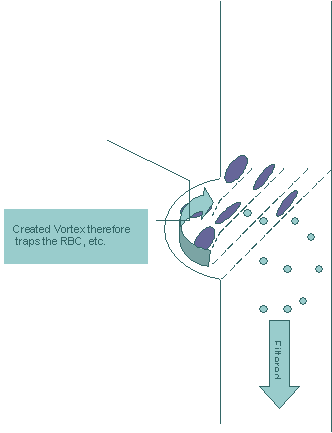
As the sample, which includes both the bacteria/viruses and the larger blood cells, is moving through the filter, the bacteria/viruses that we hope to detect will be moving through the filter due to their smaller size. The bigger cells in the sample will be blocked by the filter. However, the larger blood cells continue their movement along the tilted walls (broken lines) of the filter. At the end of the tilted plane, the space is fabricated into an oval shape. We believe vortices of the bigger cells can be created within that space, therefore trapping the bigger cells inside of that space until the rest of the sample passes through the filter – Figure 4. This effect can be utilized to prevent the re-entrance of the bigger cells into the sample. Figure 4
As can be seen in Figure 4, the liquid and the smaller particles, i.e. bacteria/virus, are passed through the filter and the bigger cells are being pushed aside and trapped by the vortex that is created in the oval area of the filter. We believe that this filter can be used to successfully filter out the bigger cells while keeping the clogging problem to a minimum.
|
|||||||||
| Electrodes |
In the previous proposal, the microchip consisted of a glass substrate with three Ti/Au electrodes insulated with a 1 mm thick layer of silicon nitride. Electrodes D and F were reference electrodes and were initially intended to electrophoretically flow sample material past the sample electrode, electrode E. (Please refer to the original proposal for detailed diagrams of the initial design.) Influenced by the new filter design, we believe that the sample will be able to flow across the detection electrode without needing electrophoretic flow. Therefore, the third electrode (F) becomes unnecessary in our improved design. Instead of using electrophoresis, we will simply use pressure to move the sample across the detection electrode. The rest of the design will stay the same: different sized areas in the silicon nitride will be etched away to expose the gold electrodes based on the size of the target to be detected. On average, bacteria organisms are on the 1mm size scale and viruses can range from 10nm-100nm. The detection electrode surface area was calculated to accommodate up to 100 bacterial or viral organisms.
|
|||||||||
| Antibody Immobilization to the Gold Detection Electrode |
Initially, we were planning to use non-specific adsorption of antibodies or Self-Assembled Monolayers (SAMs) of thioctic acid to bind antibodies to our gold detection electrode. Upon further review, we have decided to use neither of these methods to immobilize our antibodies to the detection electrode. We had several concerns regarding the prior two methods and therefore decided to use Protein A immobilization as a selective binding surface for antibody binding Concerns with regards to non-specific adsorption of antibodies to the detection electrode include: long-term stability, denaturiziation, orientation, functionality, and the ability to target these antibodies only to the detection electrode. Many of these concerns were alleviated by the use SAMs of thioctic acid. Antibodies can be coupled to these SAMs upon activation. Thioctic acid has the advantage of targeting antibody binding to the detection electrode through a covalent interaction between the thiol groups on the thioctic acid and gold. This covalent bond would also increase the stability of the antibody under a variety of conditions. The disadvantage of using thioctic acid or other similar chemistries to bind antibodies to gold surfaces are that they require several activation steps utilizing EDC and NHS in order to covalently bind the antibodies to the SAM. It has also been reported that covalent binding leads to a decrease in reactivity of the antibodies (Stefan et al., 2000) . We have therefore decided to use Protein A immobilization to the gold detection electrode as the first step of our antibody immobilization. Protein A can be immobilized onto gold surfaces either by non-specific adsorption or covalent binding. The non-specifically adsorbed Gold-Protein A complex is a highly stable complex and is believed to be Van der Waals in nature (Davis et al., 1989) . However, covalent binding of Protein A to the gold detection electrode is a much more attractive alternative, and can be achieved by genetically engineering the protein to contain cysteine residues at its C-terminus. These covalently bound Protein A molecules have been reported to have a higher IgG binding activity than physically adsorbed Protein A (Kanno et al., 2000) . In addition, the antibodies bound to these genetically engineered proteins have also been shown to have a greater antigen binding activity (Kanno, Yanagida et al., 2000) . We hope to use Protein A in our biosensor as a direct immobilization method due its natural affinity towards the Fc region of IgG antibodies (Babacan et al., 2000) . This allows us the flexibility to bind any antibody without denaturing it and to orient the antibody molecule in such a manner that will not block the antigen binding sites (Deshpande, 1996) . This method has also been shown to be functional, by its ability to bind Salmonella through the immobilization of anti-Salmonella antibodies (Babacan, Pivarnik et al., 2000) . The fact that this genetically engineered Protein A can form covalent bonds with the gold through thiol-gold interactions increases the stability of the antibody binding to the gold detection electrode and allows us to potentially target the antibodies only to the detection electrode. This method seemed to be the most logical solution for our biosensor in order to bind antibodies to our detection electrode that would be oriented, stable and functional. Babacan, S., Pivarnik, P., et al. (2000). Evaluation of antibody immobilization methods for piezoelectric biosensor application. Biosensor & Bioelectronics 15, 615-621. Davis, K. A. and Leary, T. R. (1989). Continuous liquid-phase piezoelectric biosensor for kinetic immunoassays. Anal. Chem. 61, 1227-1230. Deshpande, S. S. (1996). Antibodies: Biochemistry, structure, and function. Enzymes Immunoassays: From Concept to Product Development. NY, Chapman and Hill: 24-51. Kanno, S., Yanagida, Y., et al. (2000). Assembling of engineered IgG-binding protein on gold surface for highly oriented antibody immobilization. J. Biotechnol. 76, 207-214. Stefan, R.-I., Staden, J. F. v., et al. (2000). Immunosensors in clinical analysis. Fresenius J. Anal. Chem. 366, 659-668.
|
|||||||||
| Electronics: | We
have decided to utilize “off the shelf” electronics for a bulk of the
measurement and testing equipment for the biosensor.
We have chosen to do this because of the other complications
surrounding the fabrication of the device and the noted lack of an
electrical engineer on our team to help us with such designs.
During our searching for various components, we have discovered
that there are several commercially available miniaturized lock-in
amplifiers that could be used for the amplifier portion of the device.
In addition, we have found several reasonably priced oscillator
chips that could provide the correct frequency AC signal from a DC
(battery) source. These
components would enable us to carry out the measurement and amplification
in a small sized device. Additionally,
the components that we have selected function at low voltage and power (5
volts or less at a few milliwatts) which is absolutely feasible for a
battery powered device). The
actual signal analysis is a much more complicated endeavor, and therefore
we have decided that a laptop computer would be the easiest and most
reliable way of analyzing our output signal at this time.
With further development of such a device, however, it would be
both desirable and feasible to design a device-specific analyzer and
readout. This, however, is
beyond the capabilities of our group members at this time.
|
|||||||||
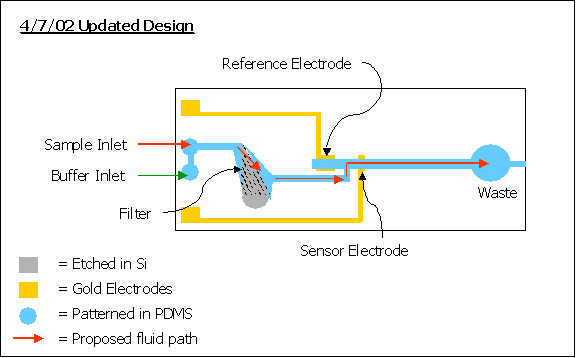
| Updated Device Layout: |
Found
below is our current design for the biosensor.
Please refer to the initial proposal for a review of the previous
design.
|
|||||||||
| III. Difficulties: | ||||||||||
| Group Dynamics |
During the course of our work, we have run into
relatively few difficulties, save the nature of our team.
While it is not the fault of any of our team members, we have had
both the joys and the difficulties afforded by a small group.
While it is easier to plan meetings and synchronize schedules,
having a small group effectively creates more work for each individual
team member. Additionally,
having a small group reduces the number of backgrounds and expertise that
is available. Adding to this,
one of our team members is also in a different physical location.
This has forced us to do much of our meeting over email and through
voice and video conferencing. While this allows us to keep in touch, it forces us to be
constantly creative in the way that we explain and exchange our thoughts
and ideas.
|
|||||||||
| Technical Difficulties | Most of the technical difficulties have been
outlined in the individual sections found above. Our greatest technical difficulties have been the filtering
issue, as well as some of the issues concerning the electronic portions of
the device. We are at a
slight disadvantage because we do not have an electrical engineer in our
group, but we are doing the best that we can with the experience of the
group members. One final area
of concern has been the end result, i.e., the final package.
Since we initially set out to design a device that is both
hand-held, portable, and low power consumption, we have a large range of
challenge in addition to our basic fabrication challenges.
Because of this, we have had to make some sacrifices with the
miniaturization of our sensor. The
most complicated part of this design is obviously the analysis equipment
which we have opted to delegate to a laptop-style computing device.
Therefore, the miniaturization of the device itself is critical to
make sure that we haven’t just designed another clunky piece of
laboratory equipment, negating the purpose of a nanofabricated biosensor.
We are currently working to solve these problems and design a
device that fits most of our initial criteria.
|
|||||||||
| IV. Summary: | ||||||||||
|
Our group has found this to be an excellent project that is well suited to our various disciplines and backgrounds. By splitting the project into sub-units, we have been able to tackle different areas based on our personal expertise. Each member has worked hard to keep the other group members “up to speed” on their portion of the project and we have had reasonable success in critically reviewing and updating our design. As can be seen from this progress report, we have made several significant changes from our initial design with the hopes of making a better and more feasible final product. While there are still some design issues that we are working on, we feel that we have a solid design to work with from this point forward. In the near future we hope to finalize some of our fabrication details, as well as to have a better electronics design for our final presentation. |
||||||||||
Team Journal
With group number of
four, each member has put a lot of effort and time into the team project. We
constantly communicate with each other regarding the project details. The
Cornell students usually discuss problems and update others with new findings
after classes. Teleconferencing, videoconferencing and online chatting are
effective ways for us to meet up with entire group because one of our group
members is at the Wadsworth Center and is not able to be physically present in
meetings. To date we have had eight of these meetings and each of them was
outlined for subsequent reference. Group meetings usually last several hours.
Because of the different backgrounds each member has, the Cornell students stay
after meetings to insure each member understands the project components and have
brainstorming time. Electronic mail has been frequently used for group
exchanging of information and meeting time scheduling as well.
Emails also allow for discussion when the group can’t find a block of
time or space at the conference facility. When each finishes partial work, we
upload the files to course website and share with other members. Detailed
descriptions of this journal is provided on the communication log and minutes
section.
|
Group
Communication Log |
||
|
Date |
Activity |
Summary |
|
2/2/02 |
|
Group introduction |
|
2/4/02 |
|
Group introduction |
|
2/6/02 |
|
Group introduction |
|
2/12/02 |
|
Introductions of individual projects |
|
2/13/02 |
|
Individual project proposals upload |
|
2/16/02 |
|
Individual project proposals upload |
|
2/17/02 |
|
Meeting scheduling |
|
2/18/02 |
Online chat |
Discussion of individual and team project |
|
2/19/02 |
|
Meeting scheduling and individual projects discussion |
|
2/20/02 |
e-Conference |
Discussion of team project presentation |
|
2/22/02 |
|
Upload final group proposal |
|
2/26/02 |
|
Team project presentation |
|
2/27/02 |
|
Project development and reference review |
|
3/1/02 |
|
Reference passing for electrode device |
|
3/6/02 |
|
Group meeting scheduling |
|
3/12/02 |
Group meeting |
Determine group strengths |
|
3/18/02 |
Group meeting |
Discussion design of fab devices |
|
3/25/02 |
|
Group meeting scheduling |
|
3/26/02 |
Group meeting |
Discussion of upcoming deadlines for the progress report |
|
3/27/02 |
Teleconference |
Design discussion of electrodes and filter development |
|
3/28/02 |
|
Reference passing for electrode device and filter design draft |
|
3/30/02 |
|
Blood sample calculation |
|
4/1/02 |
|
Group meeting scheduling |
|
4/2/02 |
|
Group meeting scheduling |
|
4/3/02 |
Group meeting |
Progress report and project design |
|
4/3/03 |
|
Reference passing for oscillator and Lock-in amplifiers |
|
4/4/02 |
|
Sensor design draft passing |
|
4/5/02 |
|
Progress report |
|
4/7/02 |
Teleconference |
Design discussion and clarifying; progress report compiled |
|
*Minutes included with date bold |
|
|
·
Feb. 18, 2002 – Group Online Meeting
- Meet online first to discuss our
individual projects and the possibility to combine our work toward the team
project. Tomas has proposed “Design and Development of a Biological Warfare
agent Biosensor” which will be applicable for any biological system for which
an antibody-antigen is available. Tom’s project was chosen and we planed to
meet again to discuss in details and prepare for the team presentation.
·
Feb. 20, 2002 – E-Video conference with
Tom, detailed meeting with Nate, Sam and Andrea
- We used the e-video conference facility in
Kimball hall and Tom explained his design and application of the biosensor
device. Tom distributed his work and powerpoint slides, and Nathan put together
of all the information for the paper work. Andrea, Nathan, and Sam will present
the team project in class and Tom is available to answer any questions during
the presentation from the Wadsworth Center.
·
March 12, 2002 – Meeting after class
- A very short
discussion for each member needed to look at the project design and reading over
spring break to put everyone on the same page.
·
March 18, 2002 – Group meeting
- Andrea came by Nate’s
lab over spring break and had a look at some of the feb devices that he made.
Andrea and Nate chatted about the project and biosensor design problems we were
facing. Another group meeting is needed ASAP for brain storming to solve the
problems.
·
March 26, 2002 – Meeting after class
-Discussed the upcoming deadlines for the progress report and the final presentation
-Discussed delegating different design components to various team members
-Sam updated Andrea and Nate on his findings on the electrical portion of the device
-Decided on a meeting time for an e-conference (chat room)
-Brought up design issues/problems with original design:
-filter
-PDMS issues
·
March 27, 2002 – Teleconference with Tom,
detailed meeting with Nate, Sam and Andrea
I.
Teleconference with Tom
-Teleconferenced with Tom for approx. 45 minutes discussing the project
-Sam filled us in on the background for the electrical measurements (impedance, etc.)
-Tom discussed the previous work done by Applied Biosystems
-Tom discussed his current problems with cell attachment to antibodies and PDMS not sticking to glass
-Group discussed possible alterations to the device:
1. change the filter from PDMS to one integrated into the device
2. possibility of attracting cells to electrodes by electric fields
3. removal of the third “electrophoresis” electrode
4. updated pumps and valves to be integrated onto device (in PDMS?)
5. miniaturization of the amplifier
6. use of silicon as substrate with SiO2 insulating layer (to fabricate easier
w/ filters on the chip instead of in PDMS)
II.
Nate, Andrea, Sam
-Discussed new filter idea:
1. integrate filter into the chip, not the PDMS
2. use diagonal filter array such as the Bob Austin lecture to filter out
larger blood material from the bacterial cells and viral agents
3. create closed waste chamber that will build pressure to prevent all of the
material from flowing past the filter
4. use PDMS as a capping material to seal the filter area, but leave
channels in the PDMS for the rest of the chip
-Andrea will do some calculations on how many of the different kinds of blood cells are in 10ul of blood and what their approximate volumes are (to give an idea of how much filtering we need to do)
-Sam will continue with the electronics design
-Nate will check into more of the fabrication details and consider some of the size requirements/layout issues
-Tom will continue with his research and will send us slides with some of his fabrication details
·
April 3, 2002
- Group meeting
I. Topics in Progress need to be
covered:
- Filter – Sam
- Nature of sampler – Andrea
Sampler may include: blood…
Air sample may be too difficult.
- Pumps – Nate
- Monolayers, etc.- Tom
- Electrodes – Sam
- Small amplifiers? - Sam
- General Nanofab – Nate & Tom
- Compile of our journal, minutes, meetings – Andrea
- Why the device has to be nano-scale – Nate
- New drawing
of the design (with new filter design and the linear flow of the sampler) –
Nate
II.
Challenges:
- Filter design
- Construction in PDMS, Si, incorporation of filter
- Attaching anti-body with cells
- Fabrication steps of incorporating anti-bodies with the devices
- Pumps
III. Changes:
- Filter
- Removal of the 3rd electrodes
- Layout –
waste
-Phone conference with all group members (Tom in Albany)
-Discussed questions about the progress report
-Went over some finer points of
the fabrication and design
-Decided on some more changes:
1. Filter to be 2mm width (to keep out platelets)
2. Filter probably can’t be 2mm deep…otherwise larger material can’t pass down the “lanes”
3. Andrea suggests using “rampart” idea, 2-step lithography to create deep lanes but narrow openings for the cells….
4. re-design some of the microfluidic layout so that only buffer passes over the larger “reference” electrode
5. pressure release outlet to be placed on waste reservoir
6. PDMS seal at the end of the “inlet” to provide septum for piercing
7. 2 syringe design, one w/ buffer, one w/ sample
8.
will inject 10ml
with sample syringe, then pump through device with buffer
-Update on the electronics:
1. have found miniaturized lock-in amplifier to be used
2. oscillator chips to be used to supply the AC current from DC battery source
3. Tom says use between 3-7kHz for frequency
4. Need to have sample electrode smaller (much smaller) than reference elec.
5.
Possibility of patterning oxide layer over the “wires” to provide
insulation
-Antibody preparation:
1. Tom suggests using Protein A as the adhesion layer for the antibodies’
2.
Protein A more generalized, but harder to put down in specific areas
-Updates on individual reports for the progress report, everyone seems like they have stuff under control
-Will
meet with Tom on Tuesday before class (Big Red Barn)
Copyright
@ Andrea Kao, Cornell University
[email protected]